 Enzyme
Catalyzed Reactions
Enzyme
Catalyzed Reactions
This page gives some theory about enzymes and
the procedures needed to extract, dilute
and use plant peroxidase. First, ensure that you
have the materials and reagents needed for these assays.
Specific procedures follow for studying the effect of [enzyme],
[Substrate], pH, inhibitors
or temperature on the rate of a peroxidase catalyzed
reaction. You can also jump to other lab
procedures in an appendix, my home
page or the addresses at the bottom.
Enzymes, as you know, are proteins
which act as catalysts in biochemical reactions. A catalyst cannot initiate
a reaction that would not happen in its absence, but it can, and does,
radically affect the reaction rate with the result that the cell can carry
out rapid and complex chemical activities at relatively low temperatures.
Most enzymes are highly specific. They tend to accelerate only one or a
group of related reactions. The result is that many different enzymes may
be present in a cell and may act simultaneously without mutual interferences.
Here we demonstrate the characteristics of enzyme catalyzed reactions by
examining peroxidase (E.C. 1. 11. 1.7).
Hydrogen peroxide (H2O2) is a common end product
of oxidative metabolism and, being a strong oxidizing agent, could prove
toxic if allowed to accumulate. To prevent this, eukaryotic cells have
enclosed the enzymes producing peroxides within a membrane-bound organelle,
the peroxisome, which is similar in size and appearance to a lysosome.
Peroxisomes also contain high concentrations of peroxidase whose function
is to reduce the peroxide to water, rendering it harmless. A variety
of electron donors are used, including aromatic amines, phenols, and
enediols like ascorbic acid.
A dye like o-dianisidine can be used as the electron donor to detect
peroxidase because its oxidized product is colored. The rate of appearance
of this colored pigment can be measured colorimetrically and is equivalent
to the rate of reaction.
H2O2+
Colorless Dye(reduced) peroxidase
>
H2O + Colored Dye(oxidized)
You can go back to the top, to
the enzyme dilution, reaction mixture preparation
or assay procedure for the [Enzyme] section, or the
closing
from here.
Materials and Reagents:
Equipment Needed:
-
Spectronic 20 photometer, set at 460 nm
-
dH2O squirt bottles and Kimwipes with each Spec 20
-
1 or 2 clean cuvettes
-
1, 5 and 10 mL pipettes with pipettors
-
100 µL pipettors and tips
-
Parafilm squares (2 cm2) - 5-10 available
-
Waring blender and blade cup assembly
-
50 mL screw-capped centrifuge tubes
-
Buchner funnel with vacuum flask and filter paper
-
Acid alcohol tub for cuvettes - 1 vol. concentrated HCl / 9 vol. ethanol
Reagents Needed:
-
30 % hydrogen peroxide (H2O2 - fresh, in refrigerator)
-
200 mL stock H2O2 - 0.10 mL 30 % H2O2
in 200 mL dH2O just prior to experiments.
-
40 g turnip or rutabaga for each enzyme preparation
-
50 mL 0.50 % w/v o-Dianisidine dye* (Sigma
#D-3127) - 0.250 g in 50 mL methanol, dissolve, filter (gravity) just before
use and store tightly capped. Add 5 mL to each of 8 test tubes capped with
Parafilm or marbles.
-
1 L 0.10 M buffer, pH 7.0 - 5.1 g Na2HPO4 + 4.9 g
KH2PO4 in 1 L dH2O.
-
0.004 M HgCl2 (F.W. 271.524) - 0.10 mL 0.02 M stock to 5 mL
dH2O, or 0.0011 g HgCl2 to 10 mL dH2O.
Label "CAUTION: POISON"
-
0.10 M KI (F.W. 166.01) - 0.166 g KI to 10 mL dH2O. (or 0.1
M CuSO4?)
-
200 mL each of 0.10 M H3PO4 (23.06 g conc./ 2 L final)
adjusted w/ NaOH to pH 2, 4, 5, 6, 8 and 10.
*CAUTION: This dye is a proven carcinogen and possibly toxic.
Avoid inhalation or contact with powder or solution.
The substrate (reactant) solution used is made by diluting
0.10 mL of 30% H2O2 to 200 mL with water. Calculate
the molarity of this solution! You will need this to calculate substrate
concentrations when plotting your data. The molecular weight of H2O2
is 34 daltons, and therefore, 34 g of H2O2/ L of
solution is a one molar concentration (= 1.0 M H2O2).
A 30% (w/v) concentration is 30 g of H2O2/ 100 mL
of solution.
Procedure: Extraction
of Peroxidase
-
Peel, wash, and cut a turnip (rutabaga, radish, horseradish root) into
1" cubes .
-
Homogenize about 40 g in 200 mL of dH2O in a blender at high
speed for 3-4 x 15 sec.
-
Clarify the extract by centrifugation (10-15,000 rpm/ 10 min.) and/or suction
filtration through Whatman #1 paper.
-
The extract may be stored under toluene for at least a week at 4°C,
if necessary.
Why would water, rather than the usual isotonic buffer,
improve peroxidase extraction?
You can go back to the top,
to the enzyme dilution, reaction mixture preparation
or assay procedure for the [Enzyme] section, or the
closing
from here.
The Effect of Enzyme Concentration
When developing an enzyme assay, one needs to adjust the enzyme
concentration so the reaction rate will be easily and accurately detected.
In addition, you must determine if the enzyme extract contains any unknown
activators (such as organic or inorganic cofactors) or inhibitors, because
these must be controlled or removed if further experiments are to be valid.
Both of these requirements can be met by assaying various enzyme concentrations
under standardized conditions and saturating levels of substrate, as given
below.
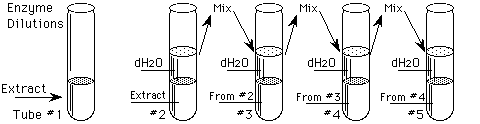
Procedure: Enzyme dilution
Prepare a stock dilution of each enzyme concentration needed as follows:
Table 1. Preparation of Enzyme Dilutions
|
Tube #
|
Enzyme Added (mL)
|
dH2O Added (mL)
|
Total Dilution
|
Total ml
Left in Tube
|
|
1
|
2.0 mL of Extract
|
0 mL
|
None = 1/1
|
2.0
|
|
2
|
2.0 mL of Extract
|
2.0
|
1/2
|
2.0
|
|
3
|
2.0 of Tube #2
|
2.0
|
1/4
|
2.0
|
|
4
|
2.0 of Tube #3
|
2.0
|
1/8
|
2.0
|
|
5
|
2.0 of Tube #4
|
2.0
|
1/16
|
4.0
|
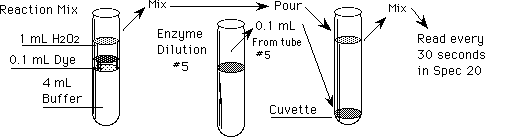
Procedure: Reaction Mixture preparation
In clean tubes, prepare 5 reaction mixtures and a reagent blank (to
adjust the Spec 20 to 0 absorbance) as indicated in Table 2 (do not
add
the enzyme!).
Table 2. Preparation of Reaction Mixtures
|
Tube #
|
mL, pH 7 Buffer
|
+ mL of Dyea
|
mL of H2O2
|
+ mL of
Enzymeb
|
from Tube #c
|
Dilution
|
|
1
|
4.0
|
0.10
|
1.0
|
|| 0.10
|
1
|
1/1
|
|
2
|
4.0
|
0.10
|
1.0
|
|| 0.10
|
2
|
1/2
|
|
3
|
4.0
|
0.10
|
1.0
|
|| 0.10
|
3
|
1/4
|
|
4
|
4.0
|
0.10
|
1.0
|
|| 0.10
|
4
|
1/8
|
|
5
|
4.0
|
0.10
|
1.0
|
|| 0.10
|
5
|
1/16
|
|
6d
|
4.0
|
0.10
|
1.0 of dH2O
|
|| 0.10
|
1
|
1/1
|
aCaution: o-dianisidine dye is carcinogenic
and
toxic. Avoid contact with your skin (wash thoroughly after use)
and notify your instructor immediately if a spill occurs.
bDo Not add the enzyme until you are ready to start the
reaction. This is time = 0!
cThese numbers refer to the enzyme dilutions in Table
1.
dThe blank contains all the reagents used in the assay except
that distilled or deionized H2O is substituted for H2O2
to prevent any reaction.
You can go back to the top,
to the enzyme dilution or reaction mixture preparation
for
the [Enzyme] section, or the closing from here.
Procedure: Reaction Rates with Enzyme Concentrations
Test how the reaction rate varies as you change the concentration of
enzyme in each reaction as follows:
-
Adjust the spectrophotometer to 0 and maximum absorbance (see appendix).
-
Set the wavelength to 460 nm.
-
With the front-left knob, set the needle to infinite absorbance with the
sample chamber empty.
-
Add enzyme to the cuvette and pour in reaction mixture #6. Cover with film
and mix gently by inversion.
-
Insert the cuvette with the blank solution (#6) into the chamber, close
the lid, and adjust the needle to 0 absorbance with the front-right knob.
-
Recheck both settings before starting the assays.
-
Save the blank solution in tube #6 and rinse the cuvette for use with the
assays; or use a matched cuvette (How?) and save the blank in its own cuvette.
-
Start the reaction by adding 0.10 mL (100 µL) of enzyme dilution
from tube #5 (= 1/16) into the cuvette and adding reaction mix #5 - this
is time = 0!
-
Cover the cuvette mouth with Parafilm®, invert several times to mix,
and insert into the sample chamber to read the absorbance at 30 second
intervals.
-
-
Read the absorbance every 30 seconds thereafter until the absorbance exceeds
1.0, or 8 minutes have passed.
-
Discard the mixture immediately (it will be orange), and thoroughly
clean
the cuvette.
-
Repeat with the other reaction mixtures and the different enzyme dilutions
(that is #4, then #3, etc.). Any reaction mixture that turns orange before
the enzyme is added will have to be remade, after the tube is thoroughly
washed.
Analysis of Data:
-
Plot the absorbance readings (y-axis) against the total time (x-axis) they
had reacted. All 5 lines, one for each [enzyme], can be plotted on the
same graph for ease of comparison.
What shape do you expect, and what do you obtain, for
these lines? How are they arranged relative to each other? Explain!
-
Now plot the absorbance readings at one minute (y-axis) against the enzyme
concentrations used. The y-axis will actually be the change in absorbance
(from 0 units) after one minute (delta A/min), which we assume is equal
to the initial velocity (V0) of the reaction. BE SURE the x-axis
is LINEAR!
What shape is the line obtained for this plot? What does
it mean?
-
Determine which enzyme dilution will give 0.6 absorbance units in 1 minute
(delta A/min = 0.6 units). This [enzyme] will be used hereafter. Why?
You can go back to the top,
to the [Enzyme], pH,
inhibitor
or temperature sections, or the closing from here.
The Effect of Substrate Concentration
A major conclusion about the mechanism of enzyme catalysis is that the
substrates bind to a specific region of the enzyme, the active site,
forming an enzyme-substrate complex prior to the reaction. This means that
since there must be a finite number of enzyme molecules, and consequently
active sites, available at any given [enzyme], substrate molecules, if
present in enough concentration, should be able to saturate these binding
sites. At this point the initial rate of the reaction (V0) will
be at a maximum (Vmax) and will not increase when the [substrate]
is increased. These principles are derived in large part from the shape
of the graph of an experiment like that below.
Procedure: Rmix Preparation for V0 vs. [S]
-
Prepare 7 reaction mixtures and a reagent blank as given in Table 3.
-
Set the Spec 20 to 0 and infinite absorbance using the blank mixture (#7)
as above.
-
Add the H2O2 and mix just before starting each reaction.
-
Add 0.10 mL of the enzyme dilution that gives change inA/min = 0.6 units
to the cuvette and pour in the reaction mixture. This is time = 0!
-
Mix and read the absorbance at 30 s and 1 minute only.
-
Rinse the cuvette!
-
Repeat steps 3.-6. for each reaction mix.
Table 3. Reaction Mixtures for V0 vs. [S]
|
Tube #
|
mL, pH 7 Buffer
|
+ mL of Dyea
|
+ mL of dH2O
|
+ mL of H2O2
|
|| + mL of Enzymeb
|
|
1
|
4.0
|
0.10
|
0.90
|
0.10
|
|| 0.10
|
|
2
|
4.0
|
0.10
|
0.80
|
0.20
|
|| 0.10
|
|
3
|
4.0
|
0.10
|
0.60
|
0.40
|
|| 0.10
|
|
4
|
4.0
|
0.10
|
0.40
|
0.60
|
|| 0.10
|
|
5
|
4.0
|
0.10
|
0.20
|
0.80
|
|| 0.10
|
|
6
|
4.0
|
0.10
|
----
|
1.0
|
|| 0.10
|
|
7c
|
4.0
|
0.10
|
1.0
|
----
|
|| 0.10
|
aCaution: o-dianisidine dye is carcinogenic and
toxic.
Avoid contact with your skin (wash thoroughly after use) and notify your
instructor immediately if a spill occurs.
bDo Not add the diluted enzyme until you are ready to
start the reaction. This is time = 0!
cThis is the new reagent blank.
Analysis of Data:
-
Calculate the molar concentration of H2O2 ([H2O2]
= [Substrate]) in each reaction mixture. Use dimensional analysis (that
is: M = mol/L, etc.).
-
Plot the absorbance readings at 1 min. (V0 = change in A/min.)
against [H2O2].
Why is the curve shaped the way it is? What does this
demonstrate?
You can go back to the top,
to the [Enzyme], [Substrate],
pH,
or temperature sections, or the
closing
from here.
The Effect of Inhibitors and Activators
Toxins and inhibitors affect everyone's daily life, and have since organisms
first began using chemical warfare to gain evolutionary advantage over
their competitors. Those that inhibit enzymes can be classed as either
irreversible, in which case active enzyme cannot be recovered once it has
been inhibited, or reversible, where the inhibitor can be physically removed
leaving the enzyme active again. This class can be further subdivided into
competitive inhibitors, in which the inhibitor binds to the active site
of the enzyme, blocking the substrate's access, or noncompetitive inhibitors,
in which the inhibitor binds elsewhere on the enzyme, changing its shape
and thus its activity. If the substrate concentration is increased relative
to inhibitor, a competitive inhibition will be overcome, while a noncompetitive
will not.
p-Aminobenzoic acid, sodium azide (NaN3), cyanide, cyclopropanone,
L-cystine, dichromate, ethylenethiourea, hydroxylamine, sulfide, sulfite,
vanadate and the divalent anions of Cd, Co, Cu, Fe, Mn, Ni and Pb are
reported to inhibit peroxidase (Horseradish; see http://www.sigma.sial.com/sigma/proddata/p8000.htm).
In addition any salts or detergents that may be introduced during extraction
or purification could potentially inhibit or activate the enzyme. Can you
design an experiment to tell which type of inhibitor each is? You will
need to determine the effective concentrations required or find them in
the literature.
Procedure:
-
Prepare two sets of reaction mixtures as in Table
3. above, but add 0.10 mL of the chosen inhibitor to each tube (CAUTION
- toxic!) in one set.
-
Do the analysis and collect the data on both sets as in the experiment
above.
-
Plot the change in A/min vs. [Substrate] on the same graph.
How has the line changed due to the inhibitor? Which
type is it?
You can go back to the top,
to the
[Enzyme], [Substrate],
inhibitor
or temperature sections, or the closing
from here.
The Effect of pH
All enzymes display a characteristic range of pH at which they are most
active. This "pH optimum" may be due to several factors involving the structure
and ionic state of the enzyme, substrate, or cofactors (see your text).
In many cases it reflects the pH of the organelle in which the enzyme is
active. What is the pH optimum for peroxidase, and the probable pH inside
the peroxisome?
Procedure:
-
Prepare a set of reaction mixtures as in Table 2., but substitute buffers
of varying pH in all but one tube and the blank. Use pH 2, 4, 5, 6, 7,
8, & 10 if possible.
-
Start each reaction with the [Enzyme] that gives change in A/min = 0.6
units only.
-
Plot V0 vs. pH (x-axis).
At what pH is peroxidase most active? Are you sure?
Did you notice anything unexpected? Reevaluate your
conclusions.
The Effect of Temperature
At low initial temperatures, increasing it increases the rates of all
reactions, whether catalyzed or not. At higher temperatures, however, proteins
denature (as in "to cook"). Since most enzymes are proteins, temperature
changes will produce something like a temperature optimum, although at
this point the protein is already being denatured.
Can you plan a properly controlled test of
the effects of temperature on the peroxidase assay? Try!
That's all for now. Again, you can jump to the
beginning,
to my home
page or the biochemistry
or non-major's
chemistry pages.
If you have questions or comments, write the:
Author of this page: Terry
Helser - helsertl@oneonta.edu
Web Coordinator: Philip S. Bidwell - bidwelps@oneonta.edu
Or return to the SUNY @ Oneonta Home Page to see where we live and work.
Last Modified on 7/6/06 
